NRTIs and NTRTIs are antiretroviral drugs which following absorption into the body must be metabolized in the affected cell in order to become active. The NRTI metabolism involves three steps, while NTRTIs require only two steps. NRTIS are analogs of DNA building blocks. NRTIs work by imitating natural DNA building blocks. When building a new viral DNA chain, reverse transcriptase enzyme binds to NRTIs instead of binding to the natural occurring DNA building blocks. Because the structure of the NRTIs, there is no allowed attachment to the next DNA building block. DNA chain growth is terminated. HIV has developed two drug resistance mechanisms to NRTIs. Mechanism number one is decreased binding of the enzyme reverse transcriptase to NRTIs. ?(The remaining mutations that have this sort of impact on the binding affinity). The second mechanism to NRTI resistance is the increased removal of the NRTI from the elongating DNA chain.
Instead of competing with naturally occurring DNA building blocks as do the NRTIs, NNRTIs bind tightly to the enzyme reverse transcriptase thereby preventing viral RNA from being converted to DNA. In contrast to the NRTIs mutations, drug resistance to NNRTIs is normally associated with mutations that are proximal to the drug binding site on reverse transcriptase. And that distortion of this binding pocket is the mechanism of resistance.
HIV1 is a virus with a high replication rate. Within infected CD4 cells the viral nucleocapsid breaks open releasing two RNA strands and essential replication enzymes such as HIV1 reverse transcriptase. It is a heterodimer with a p51 subunit and a p66 subunit. The p66 subunit contains a finger upon and a thumb region resembling a (____ hand?). Reverse transcriptase has two catalytic domains, the ribonuclease H active site and the polymerase active site. Here single stranded viral RNA is transcribed into a RNA-DNA double helix. Ribonuclease H breaks down the RNA. The polymerase then completes the remaining DNA strand to form a DNA double helix. This proviral DNA contains the genetic material of HIV1. Therapeutical suppression of viral replication slows down the decline of CD4 cells and the disease progression. Nucleoside reverse transcriptase inhibitors (NRTIs) inhibit the polymerase active site. After metabolism to non function nucleotides their incorporation causes chain termination. The non-nucleoside reverse transcriptase inhibitors (NNRTIs) form another class of powerful antiretroviral agents. They inhibit reverse transcriptase by reducing its conformational flexibility. The thumb region of reverse transcriptase is flexible. It opens and closes like a hand. Only the closed position allows transcription of RNA. The base of the thumb has a hydrophobic pocket-like binding site. This is the target of NNRTIs. Nevirapine is an important representative of this class. It does not need to be metabolized. In its native form, nevirapine binds in the pocket. This locks the thumb in the open position and prevents the transcription of RNA. Thus nevirapine stops viral replication. Due to this mechanism, nevirapine is a potent partner in the combination therapy of HIV1 infection.
Inibidores da Protease (PIs – Protease Inhibitors)
Protease inhibitors slow down HIV replication of the viruses integrated into the host cells DNA in acutely and chronically infected cells. During the maturation process of new virions, the HIV protease enzyme cuts newly produced elements of virus (____)? polyproteins into essential functional protein products. This critical process occurs as each new virion grows from the membrane of an HIV infected cell and continues after the immature virus is released from the cell. The host cell is eventually destroyed in the process. If the polyproteins are not cut the new virus fails to mature and is incapable of infecting a new cell. Protease inhibitors are able to interfere with the functioning of the native protease enzyme. They disable the enzyme before it can cleave the new viral polyprotein into its essential protein products thereby (_____)? the new virion immature and non infectious. The mechanism of resistance to protease inhibitors may be more complex than originally thought and is not as well understood as NRTI and NNRTI resistance. When PI resistance occurs, PI interference with mutant protease is no longer adequate.
The ability of HIV to infect cells is essential to its replication. Infection of a suitable host cell such as a CD4+ T lymphocyte leads to integration of the proviral DNA into the host cell genome. It now contains the genetic information for the building blocks of new HIV virus including two viral RNA strands and three viral enzymes, one of which is the HIV protease. The protease plays a key role in the formation of infectious virus. Activation of the cell induces transcription of a proviral DNA into a messenger RNA. The viral messenger RNA migrates into the cytoplasm where components of a new virus are synthesized. Some of these components have to be processed by the virus protease which cleaves longer proteins into smaller core proteins. This step is crucial to create an infectious virus. Two viral RNA strands, the viral enzymes and core proteins are assembled. This immature viral particle leaves the cell, acquiring a new (envelope?) host and viral proteins. The virus matures and becomes ready to infect other cells. Inhibition of HIV protease can stop this replication cycle. The introduction of peptidic protease inhibitors represented a milestone in treatment of HIV infection. These types of protease inhibitors bind to protease via an extensive network of hydrogen bonds resulting in the drug being attached to the active site. This blocks the action of the protease preventing HIV replication. Unfortunately mutations in HIV protease occur frequently and may limit the use of current protease inhibitors. A change in even a few amino acids can prevent these drugs binding to protease leading to broad cross resistance. Therefore there is a need for novel protease inhibitors with high potency and improved ability to overcome the diversity of mutations in the protease. Tipranavir is a novel non-peptidic protease inhibitor that displays the essential features of such an improved inhibitor. Unbound tipranavir displays the bioactive conformation more often than current protease inhibitors. Therefore less energy is required for tipranavir to adapt its overall conformation to the requirements of the binding site. Tipranavir establishes a strong network ()? energy bond interactions with conserved elements of the protease active site that cannot be mutated without the enzyme losing its function. In addition, tipranavir makes a direct hydrogen bond with the backbone atoms of the isoleucine amino acids at position 50 on each subunit of the protease. All current protease inhibitors make this interaction indirectly through a water molecule expending energy to immobilize it. The release of this water molecule by tipranavir is an energetic favorable event. The direct bond to this conserved regions results in an improved binding of tipranavir to the protease. Finally, for current protease inhibitor mutations in the protease generally weaken the bonding interactions. Tipranavir compensates for the impact of mutations in a thermodynamically unique manner by conserving and even enhancing these important contacts. As a result of these important features tipranavir retains entire ()? even against HIV strains with extensive resistance to current protease inhibitors. In summary, tipranavir has an improved ability to overcome the diversity of mutations in the protease by adopting the bioactive conformation more frequently; by estabilishing a strong network hydrogen bonds with conserved elements of the protease; By binding directly to isoleucine 50; and by compensating for the impact of mutations by enhancing important contacts. These characteristics contribute to tipranavir’s unique resistance profile and make tipranavir an innovative option for the treatment of HIV disease.
Inibidores da fusão do HIV com as células do hospedeiro
Occuring after attachment and co-receptor binding the third step in HIV cell entry, fusion, also represents a target for anti-viral drug development. One model of fusion requires gp41 to undergo extensive structural reorganization in order to destabilize the viral and cell membranes. Compounds that bind gp41 and interfere with this process have the potential to prevent HIV cell entry. Drug candidates that block gp41, known as fusion Inhibitors, are currently in clinical development.
Inibidores da Integrase
Integrase is an essential enzyme that allows HIV to integrate its proviral DNA into the host cell cromossomes. Integrase Inhibitors are on the development as a new class of anti-HIV drugs.
Nota: Existem inibidores da fusão e da integrase que são comercializados.
A droga no sangue não se equilibra rapidamente com tecidos extravasculares;
Há dois compartimentos: um central e um periférico (tecidual). O central representa o sangue e tecidos com alta perfusão. O periférico representa os tecidos em que a droga se equilibra mais devagar.
Administração em bolus:
Droga é administrada de uma vez no compartimento central (injeção intravenosa rápida);
Eliminação e transferência entre compartimentos:
Tanto a eliminação quanto a transferência entre compartimentos são processos de primeira ordem
Equação Concentração x Tempo
Administração IV em bolus – 2 compartimentos
A modelagem segue o seguinte princípio:
Taxa de variação da concentração da droga no compartimento = Taxa de entrada da droga – Taxa de saída da droga
Para o compartimento 1, assume-se que a taxa de entrada é diretamente proporcional a concentração da droga no compartimento 2, sendo a constante de proporcionalidade.
Taxa de entrada da droga (1) =
Ainda para o compartimento 1, assume-se que a taxa de saída é diretamente proporcional a concentração da droga no compartimento 1. Entretanto, temos duas formas de saída:
Taxa de saída da droga (1) =
Para o compartimento 2, fazemos de forma análoga ao que fizemos para o compartimento 1:
Taxa de entrada da droga (2) =
Taxa de saída da droga (2) =
Observe que a taxa de variação da concentração da droga em relação ao tempo para um compartimento é dada por . A modelagem para o problema em questão gera, então, um sistema de duas equações diferenciais:
(1)
(2)
Um caminho para a resolução deste sistema é o uso da transformada de Laplace. Comecemos pela equação (1):
(3)
Apliquemos, então, a transformada de Laplace à equação (2):
(4)
Substituindo (3) em (4):
(5)
Para auxiliar a resolução, fazem-se as seguintes considerações:
Logo, voltando em (5):
É possível, desta forma, usar o método das frações parciais:
(6)
Portanto, temos que:
(7)
(8)
Isolando y em (7):
(9)
Substituindo (9) em (8):
(10)
Substituindo (10) em (9):
(11)
Substituindo (10) e (11) em (6):
Finalmente, consegue-se aplicar a transformada inversa:
De uma forma mais simplificada:
E finalmente:
Referência: Leon Shargel, Susanna Wu-Pong, Andrew B.C. Yu. Applied Biopharmaceutics & Pharmacokinetics. 5th edition, 2005.
Pode ser usado nas situações em que a droga no sangue se equilibra rapidamente com tecidos extravasculares;
Administração em bolus:
Droga é administrada de uma vez no compartimento (injeção intravenosa rápida);
Processo de Eliminação:
A eliminação da droga é um processo de primeira ordem
Equação Concentração x Tempo
Adm IV em bolus
O modelo compartimental para esta situação leva a seguinte equação diferencial de primeira ordem:
onde é a concentração plasmática do fármaco e k é a constante de eliminação.
A equação pode ser resolvida por integração direta, por ser uma equação diferencial separável:
Quando o tempo varia de 0 a t, varia de a , onde é a concentração do fármaco no tempo t=0. Logo,
(1)
Determinação de parâmetros farmacocinéticos
1) A partir de dados de concentração plasmática
1.1) Determinação de k
A idéia é transformar a equação (1)em uma equação linear. Para isto, calculamos o logaritmo natural de ambos os lados da equação:
Vendo como a variável dependente e t como a variável independente, a equação acima é uma equação do tipo , sendo e . A partir de dados experimentais, os valores e podem ser determinados com o ajuste dos dados ln(concentração)-tempo a uma reta pelo método dos mínimos quadrados. O valor de k segue imediatamente, pois
1.2) Determinação de
Primeiro, deve-se determinar a concentração da droga no instante t=0:
A concentração é a razão dose por volume de distribuição:
1.3) Determinação de Cl
1.4) Determinação de
Basta considerar dois intervalos de tempo distintos e , sendo que:
,
onde é a concentração plasmática em e é a concentração plasmática em .
Temos então que:
,
e, portanto,
Mas como
Referência: Leon Shargel, Susanna Wu-Pong, Andrew B.C. Yu. Applied Biopharmaceutics & Pharmacokinetics. 5th edition, 2005.
A potenciação de longo prazo é um fenômeno onde um nível constante de estimulação pré-sináptica é convertido em uma grande saída (output) pós-sináptica. Este fenômeno desempenha papel importante na memória e no aprendizado.
Long-Term Potentiation LTP
In normal neuronal communication, input from a presynaptic neuron leads to firing in a postsynaptic neuron. Increased stimulation from the presynaptic neuron causes increased firing of the postsynaptic neuron. But in LTP, the postsynaptic neuron continues to fire at an elevated rate even after the increased stimulation has subsided.
Os dois vídeos que seguem abaixo, falam sobre modelos que tentam explicar a ocorrência deste fenômeno:
LTP mechanisms
There are two explanations for what actually occurs at the molecular level during LTP. One hypothesis suggests that the presynaptic neuron fires at abnormal rate, releasing an elevated amount of neurotransmitter. If you release more neurotransmitter, you’re gonna get a bigger response in the postsynaptic neuron. The other way is actually to, for example, modify the receptor function. So the receptors are more sensitive to the neurotransmitter. In the second model, normal amount of neurotransmitter is released, but the increased sensitivity of the receptor, causes the postsynaptic neuron to fire at an elevated rate.
Increased Receptor Sensitivity
When the neurotransmitter glutamate binds with the receptor it opens up a channel through the membrane. Electrically charged ions enter the postsynaptic neuron, which then fires an action potential. But a higher state of sensitivity occurs when an enzyme phosphorylates the protein channel. The next time a neurotransmitter binds with the receptor, more ions stream through the channel. More action potentials fire within the postsynaptic cell, passing a strong signal along to other neurons in the network.
Segue uma explicação do mecanismo de sensibilização do receptor.
1 – Moléculas de glutamato, neurotransmissor liberado pelo neurônio pré-sináptico, ligam-se a receptores NMDA na membrana do neurônio pós-sináptico;
2 – Estes receptores NMDA contêm canais transmembrânicos para Ca2+. Assim, a ligação de moléculas de glutamato a estes receptores aumenta a permeabilidade da membrana ao Ca2+.
3 – O influxo de Ca2+ ativa a proteína cinase II dependente de cálcio-calmodulina (CaMKII). Esta cinase fosforila um segundo tipo de receptor para glutamato, o AMPA. Ao ser fosforilado, o receptor AMPA torna-se mais permeável a íons Na+, diminuindo o potencial de repouso da célula. A célula fica, então, mais sensível a impulsos de entrada.
4 – Além disto, há evidências de que a atividade aumentada da CaMKII aumenta o número de receptores AMPA na sinapse.

 a constante de proporcionalidade.
a constante de proporcionalidade.


 . A modelagem para o problema em questão gera, então, um sistema de duas equações diferenciais:
. A modelagem para o problema em questão gera, então, um sistema de duas equações diferenciais: (1)
(1) (2)
(2)


 (3)
(3)

 (4)
(4)



 (5)
(5)



 (6)
(6)

 (7)
(7) (8)
(8) (9)
(9)
 (10)
(10)
 (11)
(11)







 é a concentração plasmática do fármaco e k é a constante de eliminação.
é a concentração plasmática do fármaco e k é a constante de eliminação.
 a
a 
![(ln|Cp|)]_{C_{p}=C_{p_{0}}}^{C_{p}=C_{p}}=(-kt)]_{t=0}^{t=t}](https://s0.wp.com/latex.php?latex=%28ln%7CCp%7C%29%5D_%7BC_%7Bp%7D%3DC_%7Bp_%7B0%7D%7D%7D%5E%7BC_%7Bp%7D%3DC_%7Bp%7D%7D%3D%28-kt%29%5D_%7Bt%3D0%7D%5E%7Bt%3Dt%7D+&bg=ffffff&fg=333333&s=0&c=20201002)






 como a variável dependente e t como a variável independente, a equação acima é uma equação do tipo
como a variável dependente e t como a variável independente, a equação acima é uma equação do tipo  , sendo
, sendo  e
e  . A partir de dados experimentais, os valores
. A partir de dados experimentais, os valores 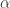 e
e  podem ser determinados com o ajuste dos dados ln(concentração)-tempo a uma reta pelo método dos mínimos quadrados. O valor de k segue imediatamente, pois
podem ser determinados com o ajuste dos dados ln(concentração)-tempo a uma reta pelo método dos mínimos quadrados. O valor de k segue imediatamente, pois






 e
e  , sendo que:
, sendo que: ,
, é a concentração plasmática em
é a concentração plasmática em  é a concentração plasmática em
é a concentração plasmática em 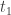 .
. ,
,






