Uma matriz monolítica é a forma mais simples e mais barata de controlar a liberação de fármacos. Para fabricá-la, um polímero ou outro material é homogeneamente distribuído com a droga por mistura da droga com o material polimérico. Os interstícios controlam a liberação da droga. O grau do controle da difusão da droga pela matriz é determinado pelas propriedades do polímero e do fármaco.
Ou a droga está totalmente dissolvida no polímero ou dispersa como partículas sólidas na matriz. A última condição prevalece quando a concentração da droga é muito maior que sua solubilidade no polímero. A cinética de liberação nestes dois estados é diferente.
Aqui será descrita a liberação para o caso da droga dissolvida na matriz. Quando a matriz entra em contato com o solvente, a droga começa a se difundir para fora dos interstícios da estrutura polimérica. A expressão matemática que rege a difusão pela matriz é uma equação diferencial parcial, a segunda lei de Fick:

Iremos analisar uma situação em que a matriz polimérica seja esférica. Em coordenadas esféricas, esta expressão é modificada para:

Considerado a difusão como radial, a equação se reduz a:

onde D é o coeficiente de difusão, C é a concentração da droga e r é a distância do centro da esfera.
Podemos então determinar as condições iniciais e de fronteira. Ao colocar a forma farmacêutica em contato com a água, a concentração da droga na superfície da esfera é zero, enquanto que a concentração dentro da esfera é única para todos os pontos. Se a esfera tem raio a, segue que:

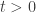


Podemos resolver este problema por separação de variáveis. Podemos primeiro fazer uma substituição que facilitará a resolução:

Perceba que:

Isolando 

Perceba também que:

Assim:

Substituindo (5) em (6) em (1), assim como (2) e (3) em (4), chegamos ao PVIF (cuja EDP é análoga a conhecida equação do calor em uma dimensão):




Podemos resolver por separação de variáveis:

Substituindo (10) em (7):


Chegamos, então, a duas EDO’s com as condições determinadas por (8) e (9)



Se multiplicarmos a equação (11) por A(r) e fizermos uma integração de 0 a a:

Usando integração por partes na primeira integral:

Chegamos a conclusão que só temos autofunções para autovalores 

Se 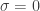

Se 

Pela condição 

Pela condição 


e, portanto:



Substituindo (14) em (13):

Logo, 
Através de (10), (15) e (16), e pelo princípio da superposição, chegamos à solução geral:

Falta agora, determinar os coeficientes 

Assim,

E, finalmente, usando (4), (17) e (18):

Referências:
C.-J Kim. Controlled release dosage form design. Technomic, Lancaster PA, 2000, 301 pp.
J. Crank. The mathematics of diffusion, Second Edition, Claremdon Press, Oxford, 1975.

